


- Download PDF
- |
- Download Citation
- |
- Email a Colleague
- |
- Share:
-
- Tweet
-
Journal of Modern Human Pathology
Volume 1, Issue 4, August 2016, Pages 35–38
Case reportOpen Access
Atypical presentation of a desmoplastic small round cell tumor in the orbit: Case report and review of the literature
-
Payal Sojitra1,
Lawrence J. Jennings2,
Rahmani Bahram3,
Yasmin C. Gosiengfiao4 and
Pauline M. Chou2,*

*Corresponding author: Dr. Pauline M. Chou, Professor, Department of Pathology & Laboratory Medicine, Ann & Robert H. Lurie Children’s Hospital of Chicago, 225 East Chicago Avenue, Box 17, Chicago, Illinois 60611-2605, USA. Tel.: 312-227-3969; Fax: 312-227-9616; E-mail: pchou@luriechildrens.org
Received 29 April 2016 Revised 30 June 2016 Accepted 16 July 2016 Published 27 July 2016
DOI: http://dx.doi.org/10.14312/2397-6845.2016-6
Copyright: © 2015 Sojitra P, et al. Published by NobleResearch Publishers. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
AbstractTop
Desmoplastic small round cell tumor (DSRCT) is a rare malignant neoplasm. Typically it occurs in young adult males and involves the abdomen or pelvis. Other primary sites are uncommon. Only one other case of orbital DSRCT has been reported in the literature. The diagnosis of DSRCTs can be extremely challenging since they often have overlapping histologic and immunohistochemical profile with other small round blue cell tumors. This is particularly true when the tumor occurs in an unusual location, and especially when it lacks the classic histologic pattern. Molecular confirmation is critical when the biopsy sample is limited. Herein, we report a case of DSRCT involving the right orbit of a 3-year-old male with immunohistological and molecular confirmation for DSRCT.
Keywords: pediatric tumors; orbit; desmoplastic small round cell tumor; qRT-PCR; sequencing; EWSR1-WT1
IntroductionTop
Desmoplastic small round cell tumor (DSRCT) is a rare malignant small round blue cell tumor. This tumor was first described by Gerald and Rosai in 1989 and got its name in 1991 [2, 2]. More than 250 case reports have been published since. Most tumors commonly originate from the serosal lining of the abdomen (frequently omentum and mesentery), thoracic cavity and pelvis (including paratesticular location). Occasionally some have been reported in such unusual sites as the head and neck, kidney, brain, lungs, ovary and orbit [3-8]. As the name implies, there is a characteristic desmoplastic stroma surrounding nests or sheets of small round blue cells. However, the desmoplastic stroma is often absent in tumors arising from viscera organs [7]. Tumor cells show a polyphenotypic immunohistochemical profile expressing mesenchymal, epithelial and neural markers in varying combination. There are many reports where either the histology and/or immunohistochemical profiles are unusual. The identification of a recurrent translocation t(11;22) (p13;q12) resulting in EWSR1-WT1 fusion transcript and selective WT1 carboxy-terminus immunoreactivity provides an accurate diagnosis in these cases [9].
Herein, we report a case of right sided orbital tumor in a 3-year-old male with an unusual presentation and histology. We present our workup strategy at arriving at the correct diagnosis using a limited immunohistochemical panel which includes WT1 (C-terminus) and molecular identification of a typical translocation of DSRCT which was confirmed by Sanger sequencing studies.
Case historyTop
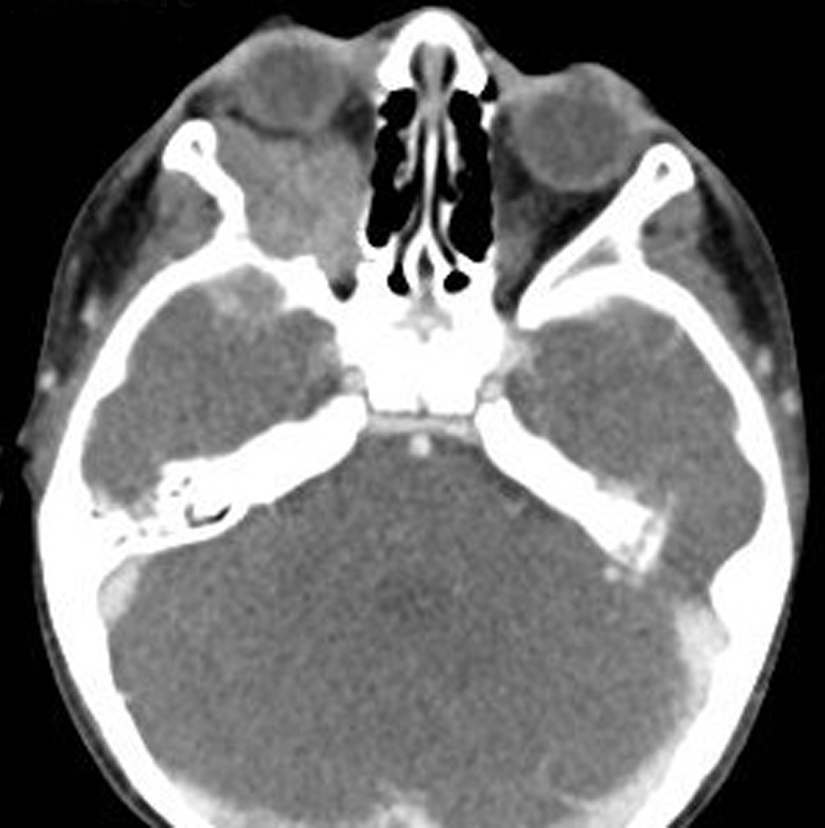
A previously healthy 3-year-old male, presented with a 2-3 week history of rightsided periorbital swelling and proptosis and one day history of right eye pain. His visual acuity was 20/20 and he had no limitation of movement in his right eye. Family history revealed a “cystic nasal neuroblastoma” in his paternal grandmother in her 40's, a paternal great grandfather with prostate cancer and a maternal great grandmother with colon cancer. Both parents were healthy. A urinalysis performed by his pediatrician to assess for nephrotic syndrome was normal. An MRI of the orbit revealed a 3.2 × 2.7 × 2.0cm lobular relatively homogeneous enhancing mass involving the inferior and lateral right orbit extending to the orbital apex with suggestion of bony invasion and disruption of the right sphenoid wing. CT scan confirmed the presence of associated erosive changes along the adjacent right sphenoid bone (Figure 1). A PET scan revealed no other sites of disease. An incisional biopsy was performed.
Materials and methodsTop
Histologic and immunohistochemical analysis
Histopathological features were assessed using Hyphanation is required and eosin–stained sections of formalin-fixed, paraffin-embedded tissue. Standard immunohistochemistry staining procedures were performed on the automated Ventana Benchmark XT system using the Ventana Optiview DAB IHC detection kit (Ventana Medical Systems, Tucson, AZ, USA). Paraffin-embedded sections were obtained and incubated with the following antibodies: WT1-C terminus (Thermo Scientific, Watham, MA, USA) at 1:100 dilution; myogenin (myf4) and BAF-47 (Cell Marque, Rocklin, CA, USA) both predilute; Myo-D1 (Dako, Carpinteria, CA, USA) predilute; EMA, AE1/AE3, S-100, NSE, CAM5.2, CD99, synaptophysin, chromogranin, WT1- N terminus (6F-H2), Desmin and Vimentin (all from Ventana, Tucson, AR, USA) predilute antibodies. Each antibody was accompanied by appropriate positive and negative controls.
Molecular studies
Real-time reverse transcription polymerase chain reaction (qRT-PCR)
Total RNA was extracted in duplicate from paraffin-embedded, formalin-fixed tissue sections. Extracted RNA was transcribed to cDNA with reverse transcriptase. The cDNA was subsequently analyzed using 3 primer sets specific to EWSR1-WT1 fusion transcripts (exon 7- exon 8, exon 8- exon 8 variants, Table 1). Also the cDNA was analyzed using primers specific to PAX3-FOXO1, PAX7-FOXO1, SYT-SSX1, SYT-SSX2, SYT-SSX4, SYT-SSX7, SYT-SSX8, EWSR1-ERG, EWSR1-FLI1 and CIC-DUX4 fusion transcripts. Products were analyzed using real-time PCR and melt curve analysis. The quality of mRNA was assessed by an independent amplification of a larger product, the ubiquitously expressed GUSB mRNA (Table 1). All primers were confirmed for specificity by BLAST search.
| Transcript | Forward | Reverse |
| DSRCTex7-ex8 99bp | CAG CCA AGC TCC AAG TCA AT | AAC CTT CGT TCA CAG TCC TTG |
| DSRCTex7-ex8 105bp | CAG CCA AGC TCC AAG TCA AT | CGA GAA AAC CTT CGT TCA CAG |
| DSRCTex8-ex8 | AGG AGG ACG CGG TGG A | CGA GAA AAC CTT CGT TCA CAG |
| GUSB 160bp | AGC CAG TTC CTC ATC AAT GG | GGT AGT GGC TGG TAC GGA AA |
| GUSB 171bp | AAA CGA TTG CAG GGT TTC AC | CTC TCG TCG GTG ACT GTT CA |
Note: All primers are shown in the 5’ to 3’ direction. All primers include M13 sequences on the 5’ end (TGT AAA ACG ACG GCC AGT on each Forward primer and CAG GAA ACA GCT ATG ACC on each reverse primer).
Sanger dideoxy sequencing
Sanger sequencing assay was used to confirm the qRT-PCR results. To remove unused dNTPs and primers left over from the qRT-PCR reaction, 10µl of amplicon was transferred to a new PCR tube and 1×l of ExoSAP-IT® (Affimetrix, Santa Clara, CA) was added. The amplicon clean-up was performed following ExoSAP-IT® manufacture instructions. 2×l of the cleaned product was used for sequencing reactions with the Big Dye Terminator v1.1 Cycle Sequencing kit (Life Technologies, Carlsbad, CA). Bidirectional sequencing was performed using the M13 sequencing primers (forward primer 5'–TGT AAA ACG ACG GCC AGT–3' and reverse primer 5'–CAG GAA ACA GCT ATG ACC–3') in an ABI Prism 3130 xl automatic sequencer (Life Technologies, Carlsbad, CA). Briefly, the forward and reverse M13 primers are added to the cleaned amplicon product in separate wells and cycle sequenced. Data were analyzed by Sequencing Analysis 5.1.1 (Life Technologies, Carlsbad, CA).
ResultsTop
Gross and microscopic features
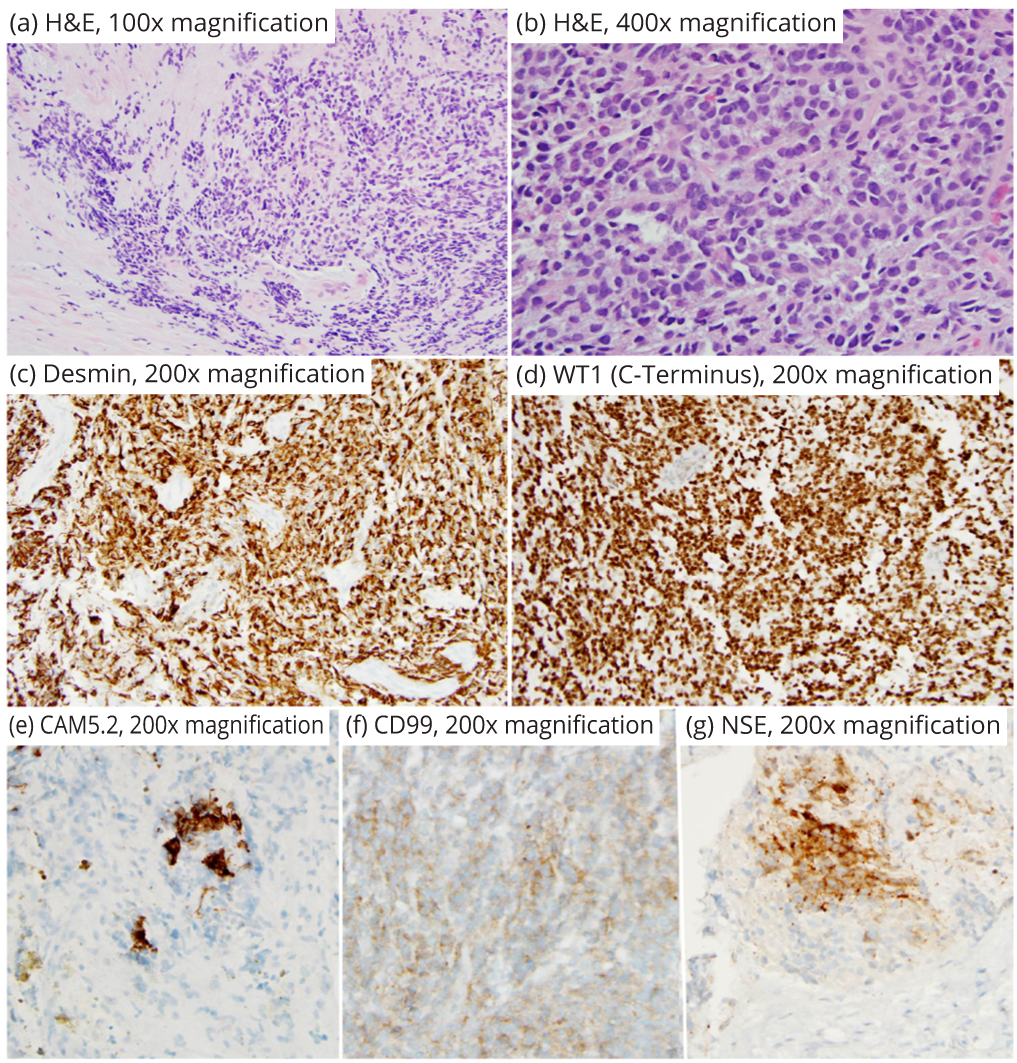
The biopsy specimen consisted of multiple fragments of tan to red glistening tissue measuring in aggregate 1.0 × 0.5 × 0.2cm. On microscopic examination, the tumor showed a variable architectural pattern with sheet-like and nested appearance separated by thin fibrovascular somewhat myxoid stroma. The tumor cells are composed of primitive round to ovoid cells with scant cytoplasm. Focally tumor cells show a spindled morphology. The nuclei are monotonous with fine chromatin, rare nucleoli and minimal pleomorphism. Specifically the tumor lacked prominent desmoplasia (Figure 2a, b).
Immunohistochemical staining pattern
The tumor cells showed a distinct dot-like cytoplasmic immunoreactivity for desmin (Figure 2c) and a diffuse nuclear reactivity for the C- terminus of WT1 (Figure 2d). It was also strongly but focally positive for CAM5.2 (Figure 2e), weakly for CD99 (Figure 2f) and focally positive for NSE (Figure 2g). Tumor cells were negative for myogenin, Myo-D1, EMA, Cytokeratin (AE1/AE3), synaptophysin, chromogranin and WT1 (N-terminus) with retention of nuclear staining for BAF47 (INI1 gene).
qRT-PCR and Sanger sequencing findings
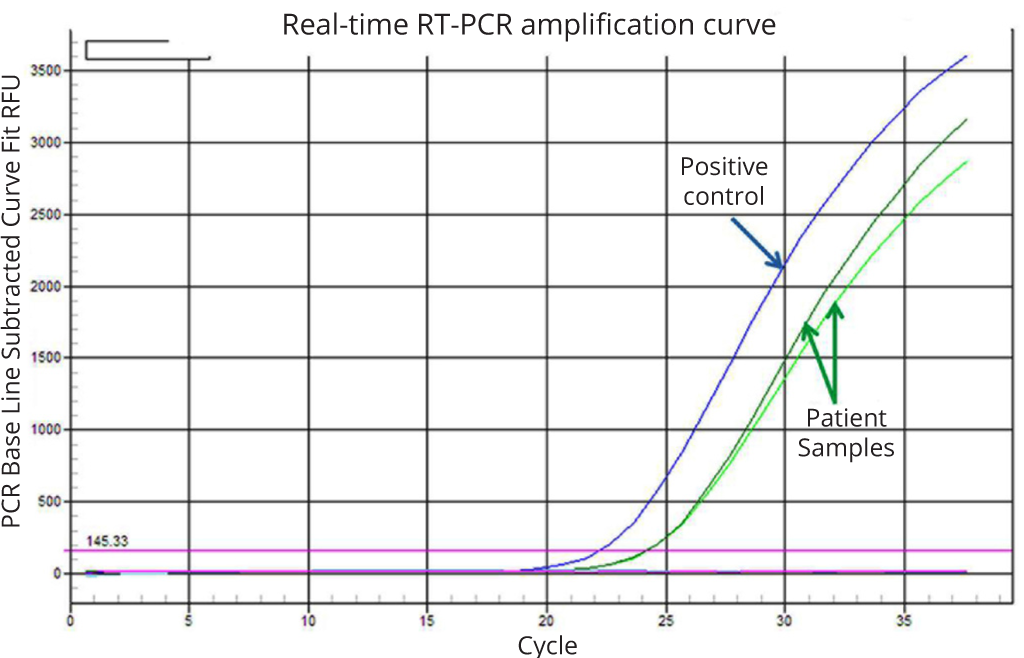
qRT-PCR showed tumor cells were positive for the fusion transcript for EWSR1-WT1 and negative for the rest of the targets (Figure 3).
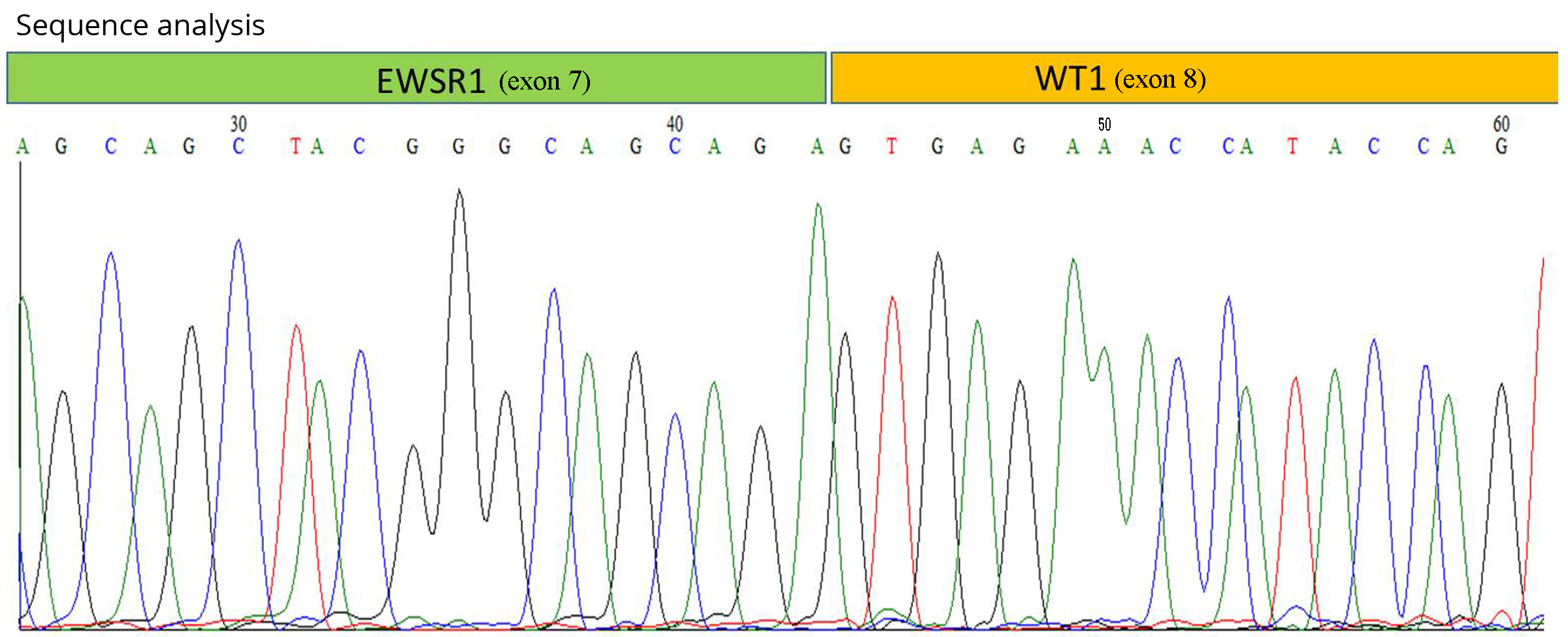
The results were confirmed utilizing Sanger sequencing assay which matches the sequence from exon 7 from EWSR1 and exon 8 from WT1 (Figure 4).
DiscussionTop
Primary round cell tumors of the orbit are rare and the diagnosis could be challenging. The differential diagnosis in our patient’s tumor is extensive like any other small round blue cell tumor, and includes leukemia/lymphoma, neuroblastoma, rhabdomyosarcoma, Ewing sarcoma and malignant rhabdoid tumor. Rhabdomyosarcoma or neuroblastoma was the primary clinical consideration based on location and patient’s age. However, though the tumor cells were immunoreactive for desmin, they were non-immunoreactive for myogenin and Myo-D1. Also the diffuse desmin immunoreactivity and negative neural markers would be unusual for either neuroblastoma or Ewing sarcoma. Ewing sarcoma is usually considered in older patients (in the second or third decade) but it can occur in any age group. This tumor historically is challenging as it can present with a diverse morphologic and immunohistochemical profile [10]. As well, FISH for EWSR1 translocation would be positive in both Ewing sarcoma and DSRCT [11]. In our case, the dot-like diffuse immunoreactivity for desmin and the immunoreactivity for keratin led us to perform additional stains which included the C-terminus for WT1 gene (positive) and BAF 47 (INI gene) (which was retained). Thus with this immunoprofile, we ruled out malignant rhabdoid tumor in favor of DSRCT. Molecular studies subsequently helped confirm the diagnosis as EWS-WT1 fusion transcript is a distinctive genetic hallmark widely considered diagnostic [9].
Generally DSRCTs have characteristic gross, histologic and immunophenotypic features, and are not difficult to recognize when the tumor presents in the abdomen or any serosal based locations. But DSRCT can present in unusual locations and may exhibit atypical histologic (spindle cell or rhabdoid) and/or immunohistochemical features causing a serious diagnostic challenge [12, 13]. To our knowledge, this is only the second case reported in the orbit. The other case report of DSRCT in this location described by Yoon et al was in a 32-year-old patient [8]. The hallmark morphologic pattern of DSRCT is that of nests or sheets of small round cells embedded in abundant desmoplasia [2]. Development of this stroma has been attributed to platelet derived growth factor-α (a potent fibroblast growth factor) which is induced by the EWSR-WT1 chimeric transcript [14]. However, others have found an inverse relationship between PDGF expression and tumor desmoplasia [15]. At present, the biologic basis or the pathophysiology of this tumor is poorly understood. Recent studies seem to indicate that the product of an aberrant transcription factor modulates the expression of genes that overlap with those generally regulated by WT1 [14].
It is evident that the case described herein is not typical of DSRCT both clinically and morphologically. The presence of this specific translocation is undeniable. No other tumor thus far, has been reported to harbor this fusion transcript except in a hybrid tumor (with features of leiomyosarcoma and DSRCT) [12] and one other cauda equina tumor with smooth muscle differentiation [16]. The latter tumor has prominent rosette formation and positive CD99, morphologic features that overlap with PNET [16]. Additionally multiple copies of EWSR1-WT1 and/or WT1-EWSR1 fusions were reported in 2 cases of DSRCT [17], and another tumor with a t(1;15) in addition to the typical EWS-WT1 translocation was found in a DSRCT with rhabdoid features [13].
It is unclear whether our tumor should be classified as a variant of DSRCT with unusual histologic and clinical features or an undifferentiated sarcoma with distinctive EWSR1-WT1 fusion transcript. However, as the tumor exhibits the classic genetic hallmark, at present it most likely belongs to the group of DSRCT with unusual morphologic feature.
The diagnosis of this rare tumor is challenging. Given the atypical presentation, uncharacteristic morphology and immunohistochemical profile, it is necessary to use ancillary molecular methods for accurate diagnosis.
ConclusionTop
Our patient is doing well so far. He had just completed therapy with no evidence of disease 15 months after his original diagnosis. He had minimal response to the initial chemotherapy regimen following the COG protocol AEWS1031 which included vincristine, doxorubicin and cyclophosphamide. This was then switched to alternative treatment with irinotecan/ temozolomide. He underwent proton (pencil beam) radiation and surgical resection for local control at another institution. Per report, the surgeon was able to resect the residual mass in its entirety with negative margins while sparing his globe. He has since been seen by Ophthalmology at our institution.
Conflicts of interest
The authors declare that they have no potential conflicts of interest which would warrant disclosure.
ReferencesTop
[1]Gerald WL, Rosai J. Case 2. Desmoplastic small cell tumor with divergent differentiation. Pediatr Pathol. 1989; 9(2):177–183.Pubmed
[2]Gerald WL, Miller HK, Battifora H, Miettinen M, Silva EG, et al. Intra-abdominal desmoplastic small round-cell tumor. Report of 19 cases of a distinctive type of high-grade polyphenotypic malignancy affecting young individuals. Am J Surg Pathol. 1991; 15(6):499–513.Article Pubmed
[3]Slomovitz BM, Girotra M, Aledo A, Saqi A, Soslow RA, et al. Desmoplastic small round cell tumor with primary ovarian involvement: case report and review. Gynecol Oncol. 2000; 79(1):124–128.Article Pubmed
[4]Mihok NA, Cha I. Desmoplastic small round cell tumor presenting as a neck mass: a case report. Diagn Cytopathol. 2001; 25(1):68–72.Article Pubmed
[5]Finke NM, Lae ME, Lloyd RV, Gehani SK, Nascimento AG. Sinonasal desmoplastic small round cell tumor: a case report. Am J Surg Pathol. 2002; 26(6):799–803.Article Pubmed
[6]Syed S, Haque AK, Hawkins HK, Sorensen PH, Cowan DF. Desmoplastic small round cell tumor of the lung. Arch Pathol Lab Med. 2002; 126(10):1226–1228.Article Pubmed
[7]Rao P, Tamboli P, Fillman EP, Meis JM. Primary intra-renal desmoplastic small round cell tumor: expanding the histologic spectrum, with special emphasis on the differential diagnostic considerations. Pathol Res Pract. 2014; 210(12):1130–1133.Article Pubmed
[8]Yoon M, Desai K, Fulton R, Bucci K, Russin M, et al. Desmoplastic small round cell tumor: a potentially lethal neoplasm manifesting in the orbit with associated visual symptoms. Arch Ophthalmol. 2005; 123(4):565–567.Article Pubmed
[9]Lae ME, Roche PC, Jin L, Lloyd RV, Nascimento AG. Desmoplastic small round cell tumor: a clinicopathologic, immunohistochemical and molecular study of 32 tumors. Am J Surg Pathol. 2002; 26(7):823–835.Article Pubmed
[10]Folpe AL, Goldblum JR, Rubin BP, Shehata BM, Liu W, et al. Morphologic and immunophenotypic diversity in Ewing family tumors: a study of 66 genetically confirmed cases. Am J Surg Pathol. 2005; 29(8):1025–1033.Article Pubmed
[11]Romeo S, Dei Tos AP. Soft tissue tumors associated with EWSR1 translocation. Virchows Arch. 2010; 456(2):219–234.Article Pubmed
[12]Alaggio R, Rosolen A, Sartori F, Leszl A, d'Amore ES, et al. Spindle cell tumor with EWS-WT1 transcript and a favorable clinical course: a variant of DSCT, a variant of leiomyosarcoma, or a new entity? Report of 2 pediatric cases. Am J Surg Pathol. 2007; 31(3):454–459.Article Pubmed
[13]Liang L, Tatevian N, Bhattacharjee M, Tsao K, Hicks J. Desmoplastic small round cell tumor with atypical immunohistochemical profile and rhabdoid-like differentiation. World J Clin Cases. 2014; 2(8):367–372.Article Pubmed
[14]Gerald WL, Haber DA. The EWS-WT1 gene fusion in desmoplastic small round cell tumor. Seminars in Cancer Biol. 2005; 15(3):197–205.Article Pubmed
[15]Zhang PJ, Goldblum JR, Pawel BR, Pasha TL, Fisher C, et al. PDGF-A, PDGF-R beta, TGFβ3 and bone morphogenic protein-4 in desmoplastic round cell tumors with EWS-WT1 gene fusion product and their role in stromal desmoplasia: an immunohistochemical study. Mod Pathol. 2005; 18(3):382–387.Article Pubmed
[16]Ud Din N, Pekmezci M, Javed G, Horvai AE, Ahmad Z, et al. Low-grade small round cell tumor of the cauda equina with EWSR1-WT1 fusion and indolent clinical course. Hum Pathol. 2015; 46(1):153–158.Article Pubmed
[17]La Starza R, Barba G, Nofrini V, Pierini T, Pierini V, et al. Multiple EWSR1-WT1 and WT1-EWSR1 copies in two cases of desmoplastic round cell tumor. Cancer Genet. 2013; 206(11):387–392.Article Pubmed
 Copyright
© 2012-2025 NobleResearch Group. All Rights Reserved
Copyright
© 2012-2025 NobleResearch Group. All Rights Reserved