



- Download PDF
- |
- Download Citation
- |
- Email a Colleague
- |
- Share:
-
- Tweet
-
Journal of Cancer Research & Therapy
Volume 3, Issue 4, May 2015, Pages 52-55
Case report Open Access
Malignant solitary fibrous tumor of the sacrum: A case report
-
Rogozhin DV1,2,*
 ,
Konovalov DM1,2,
Vanel D3,
Campanacci L3,
Boulytcheva IV1,
Talalaev AG1,2,
Roshin VY1,2,
Ektova AP1,2,
Bogorodickiy YS1,
Skapenkov IN1 and
Strikov VA1
,
Konovalov DM1,2,
Vanel D3,
Campanacci L3,
Boulytcheva IV1,
Talalaev AG1,2,
Roshin VY1,2,
Ektova AP1,2,
Bogorodickiy YS1,
Skapenkov IN1 and
Strikov VA1
*Corresponding author: Dr Dmitry Rogozhin, Pathology, Russian Children's Research Hospital, 117997, Leninsky Prospect, Moscow, Russia (Russian Federation). E-mail: pathol.777@mail.ru
Received 3 January 2015 Revised 2 March 2015 Accepted 9 March 2015 Published 18 March 2015
DOI: http://dx.doi.org/10.14312/2052-4994.2015-7
Copyright: © 2015 Rogozhin DV, et al. Published by NobleResearch Publishers. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
AbstractTop
The solitary fibrous tumor (SFT) is a rare ubiquitous mesenchymal neoplasm of fibroblastic type, which shows a prominent hemangiopericytoma-like branching vascular pattern. Approximately 40% of solitary fibrous tumors are found in subcutaneous tissue, while others are observed in deep soft tissue of extremities or in the head and neck region, thoracic wall, mediastinum, retroperitoneum and abdominal cavity. Extremely rare the SFT arise as a primary bone lesion. We have reported the first case described in literature of the primary sacrum malignant SFT in a 6-year-old child.
Keywords: solitary fibrous tumor; mesenchymal neo-plasm; children; sacrum
IntroductionTop
The solitary fibrous tumor (SFT) is a rare ubiquitous mesenchymal neoplasm of the fibroblastic type, which shows a prominent hemangiopericytoma-like branching vascular pattern [1, 2]. It was first observed to arise from the pleura and occur most commonly in the thoracic cavity. However, it is now recognized that this rare tumor can occur throughout the body [5]. Approximately 40% of solitary fibrous tumors are found in subcutaneous tissue, while others are observed in deep soft tissues of the extremities or in the head and neck region, thoracic wall, mediastinum, retroperitoneum and abdominal cavity. Other locations described include meninges, spinal cord, salivary gland, lung, thyroid, and liver [6], the gastrointestinal tract, adrenal glands, kidney, urinary bladder, prostate, spermatic cord, testis, nasal turbinate [10] and skin [2]. SFT is most commonly found in adults and affects both sexes equally [7]. SFT rarely arises as a primary bone lesion. We have reported on the first case described in the literature of a primary sacrum malignant SFT in a 6-year-old child.
Case reportTop
The 6-year-old male was admitted to the department of oncology with complaints of constipation lasting about 6 months, episodes of urinary retention, painful urination. Ultrasonography was performed and revealed a round-shaped tumor with smooth contours, homogeneous echo density, size 10x12, 8x8, 1 cm, with a large number of randomly distributed vessels. Rectum was squeezed between the urinary bladder and the tumor. The child had bilateral pyelectasis as a result of a ureteral obstruction.
CT investigation: the massive tumor was observed in the retroperitoneum with clear irregular contours and inhomogeneous structure. It actively accumulated contrast from 25-30 to 160-170 HU. The size of the tumor was 12.3x10x15.8 cm. We suspect that the origin of tumor mass was in the sacrum, causing it significant destruction (Figures 1A, B). The same lesion was found in coccygeal vertebrae. Rectum, sigmoid and urinary bladder were compressed by the tumor. The tumor blood supply was implemented through the internal iliac artery branches. Sufficiently larger arteries and veins were found at the periphery of the tumor.
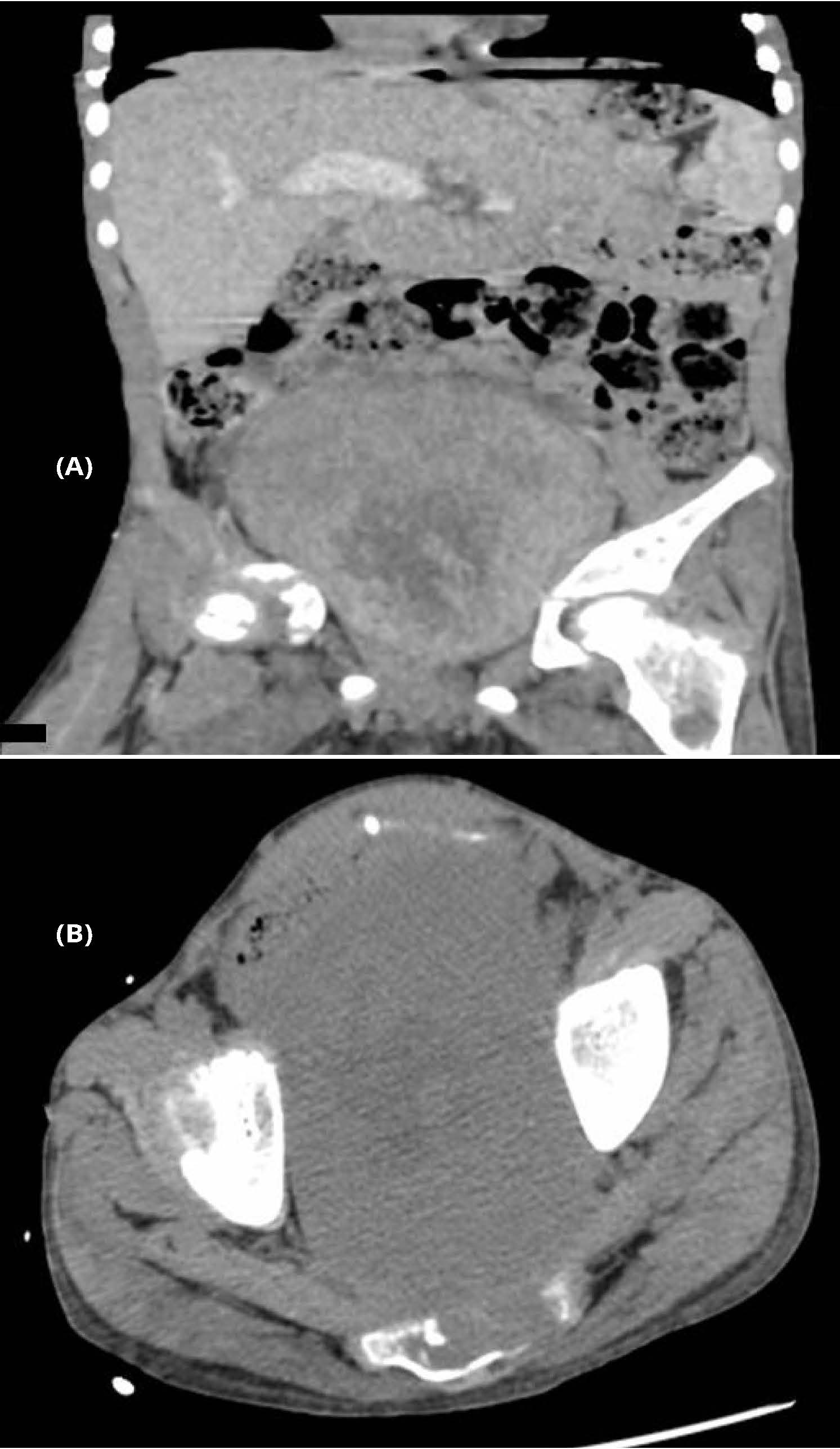
Figure 1A, B Tumor mass was found in the sacrum, causing it significant destruction. Rectum, sigmoid and urinary bladder compressed by the tumor. Palpation of abdomen revealed a large firm painless mass, with an extensively developed venous network of the anterior abdominal wall.
An open biopsy was performed: yellowish-pink tissue fragments of a soft consistency were obtained; the total volume of biopsy tissue was 2x1, 8x0, 5 cm. The tumor appeared to be hypercellular, consisting of ovoid to spindle-shaped neoplastic cells with pale vacuolated cytoplasm and indistinct borders between cells (Figure 2). The nuclei have dispersed chromatin, small nucleoli. The tumor cells tended to create multidirectional bundles. From area to area one could reveal large cells with minimal cytological atypia. The mitotic activity was 4 mitoses per 10 high power fields (HPF). In the adjacent adipose tissue invasive tumor growth was detected (Figure 3). Noteworthy: a large number of thin-walled branching hemangiopericytoma-like vessels have been observed in the tumor without prominent hyalinization around them (Figure 4).
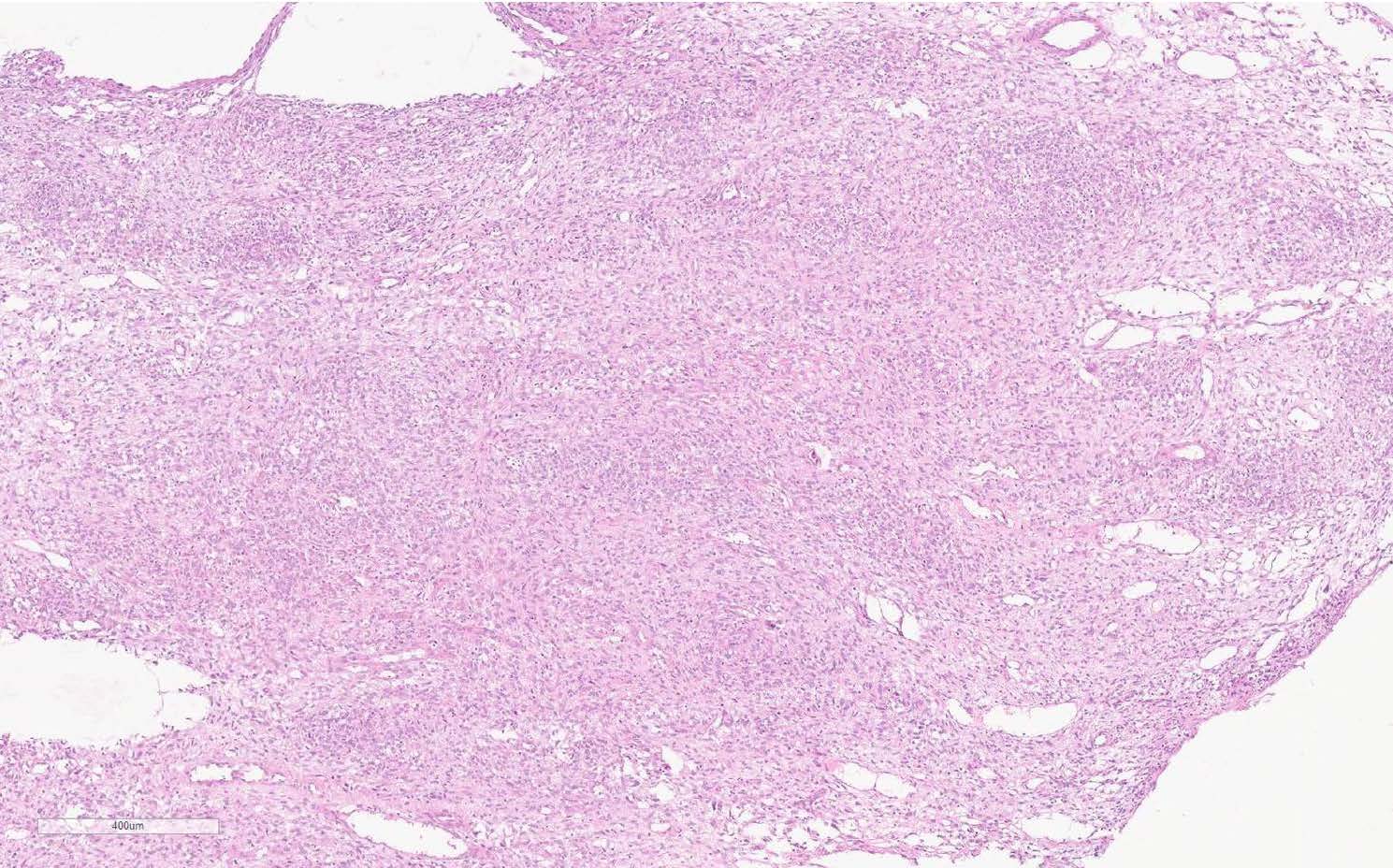
Figure 2 Histologic view of the tumor. The thin-walled vessels may be seen (H&E, scan), x50.
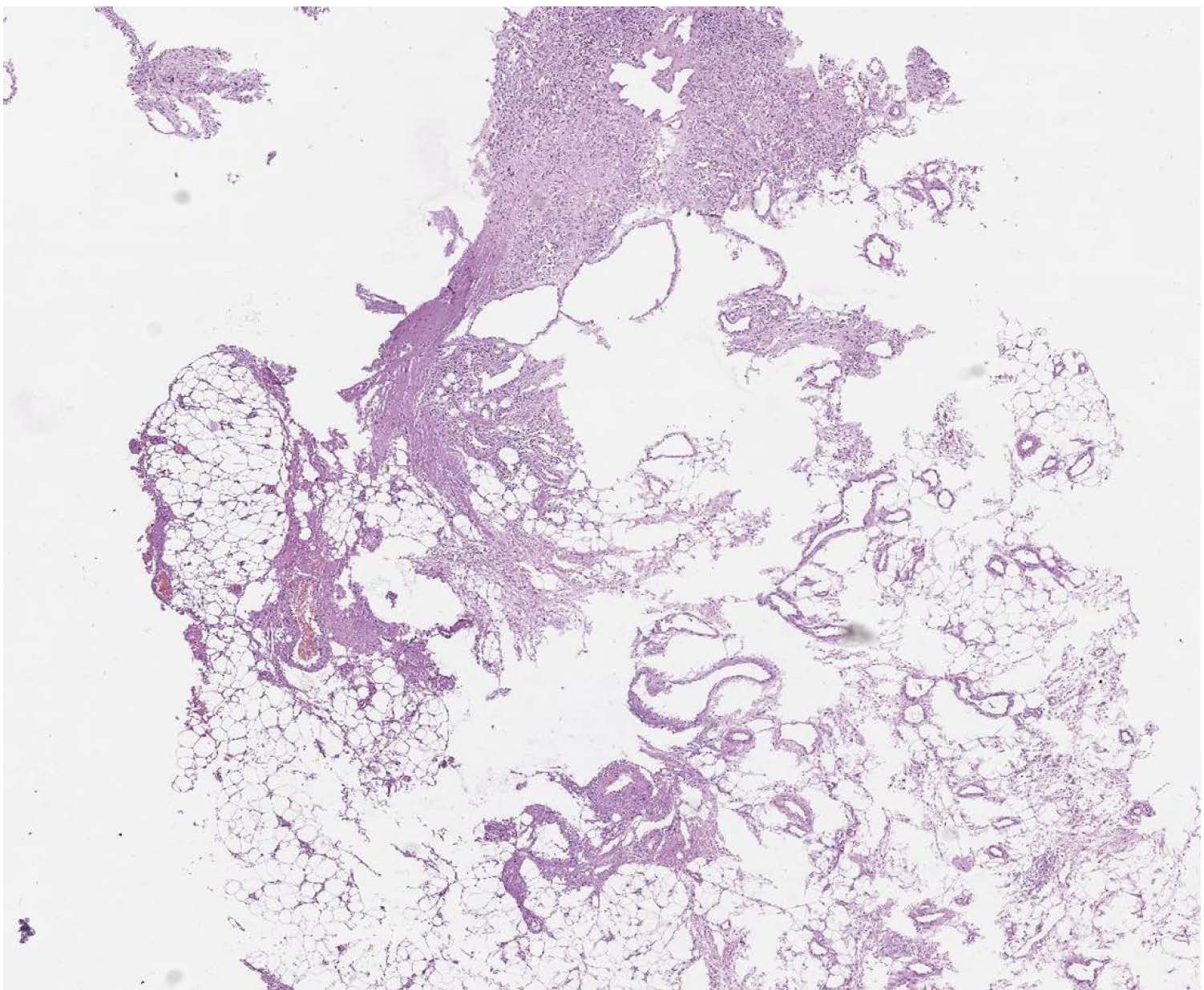
Figure 3 The invasive growth in adjacent adipose tissue, (H&E, scan), x30.
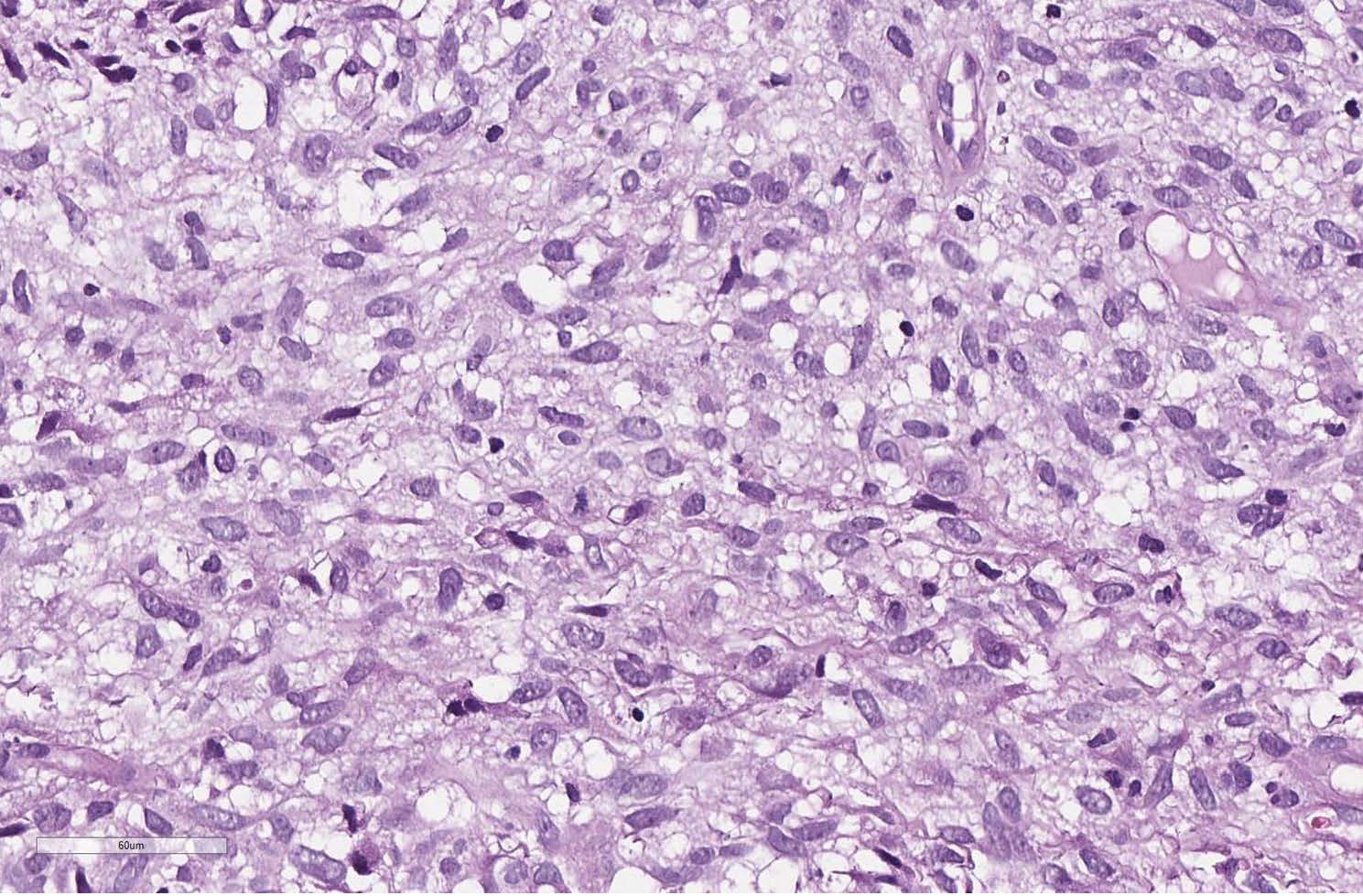
Figure 4 The tumor cells are ovoid to spindle-shaped with pale vacuolated cytoplasm having indistinct borders. Nuclei have dispersed chromatin, small nucleolei, (H&E, scan), x200.
Immunohistochemically, the tumor cells showed strong positivity for CD34, CD99, EGFR and vimentin (Figures 5, 6). Negative positivity was observed with desmin, S-100 protein, SMA, myogenin, panCK AE1/AE3, CD57, CD31, factor VIII, CD68, EMA and bcl-2.
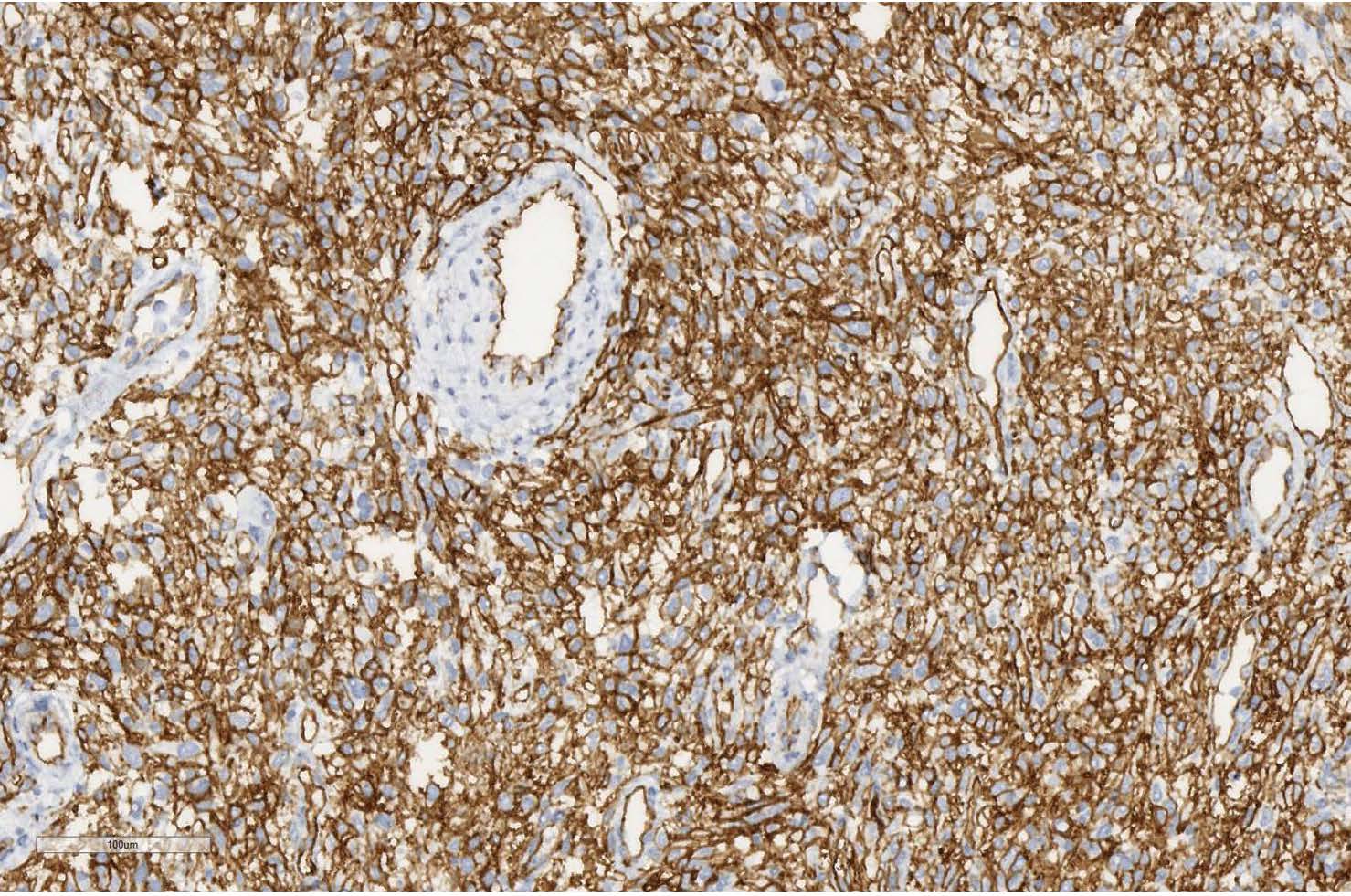
Figure 5 Positive immunostaining with CD34 (scan), x150.
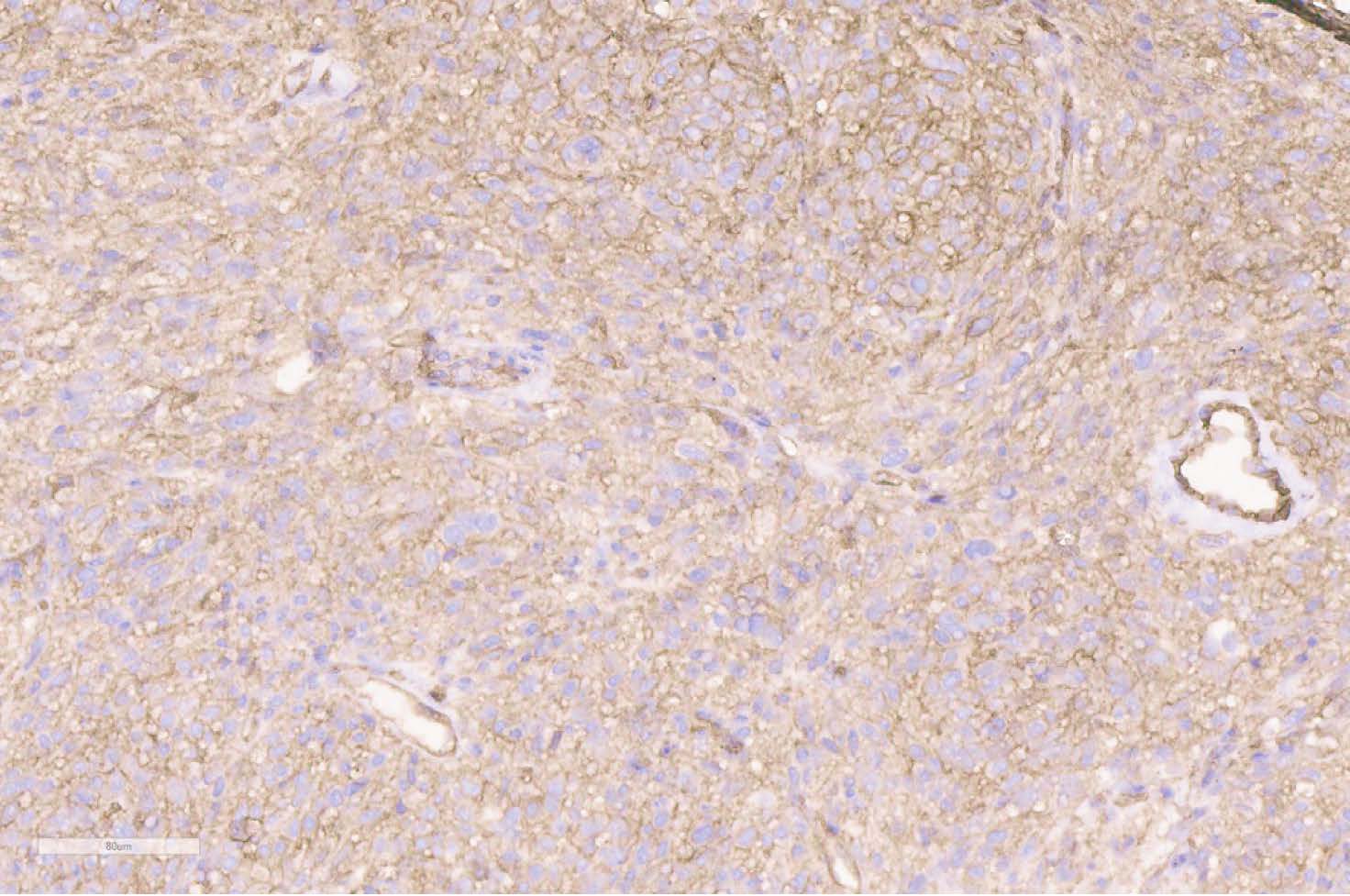
Figure 6 Positive immunostaining with CD99 (scan), x150.
Cytogenetic investigation (FISH) has excluded the EWSR1 rearrangements. The management of this patient includes bilateral nephrostomy, and chemotherapy on the first step (CWS 2009, 3 blocks) without sufficient clinical effect. After that twice super selective embolization was performed, but finally it was not effective due to rich vascularization and a lot of vascular collaterals in the tumor. Next step was target therapy by erbitux (cetuximab) with irinotecan, and last hospitalization for sigmostomia because of rectum compressing and bleeding during defecation with symptoms of anemia. After all complex of treatment procedures there are no significant points for positive clinical effect.
The final diagnosis of malignant SFT was made based on the large size of the tumor (12.2 cm in greatest diameter), the destruction of the sacrum, and the coccyx, and the involvement of other anatomical structures (rectum and sigmoid, urinary bladder and ureters), hypercellular lesion, mitotic activity (4 to 10 HPF), presence of the minimum neoplastic cell atypia, invasive growth in adjacent adipose tissue, positivity for CD34, CD99 and vimentin.
DiscussionTop
SFT is a rare mesenchymal tumor that may occur elsewhere in the body. One of the unusual locations includes the bones, especially in children. The majority of SFTs are histologically benign, but the behavior of this tumor can be unpredictable. About 10% behave aggressively. Local or distant recurrence can occur many years after primary resection [2].
The corresponding clinical symptoms appear only when the tumor has grown to a certain size, or when vital structures are involved [6]. According to the current World Health Organization (WHO) classification criteria, SFT may identify as malignant if it is a hypercellular lesion, showing increased mitotic figures (>4 mitoses per 10 HPF), variable cytological atypia, tumor necrosis, and/or infiltrative margins. Mitotic rate seems to be the most valuable prognostic factor. But malignant SFT may show cytological atypia in the absence of mitoses or necrosis [2]. Rare cases show abrupt transition from conventional benign-appearing SFT to high-grade sarcoma, likely representing the dedifferentiation. Similar to other dedifferentiated sarcomas, abrupt transition between low-grade and high-grade areas are typically observed with loss of CD34 positivity [9].
Due to overlapping histological features, differential diagnosis of SFT from other soft tissue bone lesions can be difficult [8]. For instance, hemangiopericytoma is closely related, if not identical, to SFT. Focal hemangiopericytomatous areas may be seen in several sarcomas, including fibrosarcoma and osteosarcoma [3]. Therefore, performance of immunohistochemical staining is necessary in order to rule out other tumors. Tumor cells in SFTs are characteristically immunoreactive for CD34 (90-95% of cases), 20-35% of tumors show variable positivity for EMA and SMA, 30% express bcl-2 and 70% of tumors are CD99 positive. The high sensitivity of CD34 for SFTs has resulted in a more accurate and consistent diagnosis of the entity, undoubtedly accounting for the increasing number of SFTs now diagnosed at extrathoracic sites [7]. There have been occasional reports of focal and limited reactivity for S-100 protein, keratins and/or desmin [2]. Some authors reported about negative reaction with SMA, desmin, pan-cytokeratin and S-100 protein in SFT [5]. However, these markers are frequently overexpressed by other soft tissues tumors, including hemangiopericytoma, synovial sarcoma (SS) and peripheral nerve sheath tumor (PNST).
Classic hemangiopericytoma consists of tightly packed round to fusiform cells with indistinct cytoplasmic borders, which are arranged around an elaborate vasculature. The vessels form a continuous, ramifying vascular network that exhibits striking variation in caliber. Typically, the dividing sinusoidal vessels have a “staghorn” or “antler-like” configuration. In contrast, SFT consist of principally spindle cells. A characteristic feature of the lesion is a focal striking hyalinization. Many hemangiopericytomas express CD34 but usually in a smaller percentage of cases and to a lesser degree than SFTs [7]. Reticulum stains may highlight occult blood vessels [3].
Among other tumors on differential list with SFT and its malignant counterpart is SS, especially spindle cell monophasic variant. The tumor cells in SS shows at least a focal expression of EMA in nearly all cases, whereas focal positivity for keratins (7, 8, 18 and 19) is found in 70-80% of cases. SFTs does not express these markers. More over, SS is characterized by the t(X;18)(p11;q11) translocation, which is found exclusively in this tumor [2].
In our case we have to rule out an atypical spindle cell variant of Ewing sarcoma (ES) [11]. But in case of ES the tumor cells do not express the CD34 and up to 30% show some positivity for keratin. Approximately 85% of ES have a somatic reciprocal chromosomal translocation t(11;22)(q24;q12), that fuses EWSR1 to FLI-1 to generate the EWSR1-FLI-1 oncoprotein. In other cases, alternate translocations fuse EWSR1 to other ETS family members [2].
PNST is another soft tissue tumor that may occur in bones and can be confused with SFT. Immunohistochemically, the tumor cells of PNST are diffusely or focally positive for S-100 protein, but negative for CD34, desmin and EMA [6]. Melanotic schwannomas of bone can show focal positivity for melanoma markers (Melan A, HMB-45) [4].
Some SFTs can contain giant multinucleated stromal cells and pseudo-vascular spaces, formerly known as “giant cell angiofibroma” [2]. In these cases it should be differentiated with solid variant of aneurismal bone cyst (ABC). Grossly the lesion may be completely solid. Microscopically, a spindle cell proliferation with loose arrangement of the cells is found. Very characteristically, the lesion shows abundant reactive new bone formation, with prominent osteoblastic activity similar to that of heterotopic ossification [3]. The spindle cells of ABC do not react with CD34.
In fibroblastic subtype of conventional osteosarcoma the malignant cells are usually spindled, less frequently epithelioid, and often, but not always demonstrate severe cytological atypia. The tumor cells are associated with extracellular collagen, which can be extensive, and often arranged in a storiform pattern. Cells with fibrillar eosinophilic cytoplasm have myofibroblastic differentiation [2]. In classical osteosarcoma, usually, there are no prominent thin-walled vascular spaces. The neoplastic cells in osteosarcoma have a broad immunoprofile that lacks diagnostic specificity. Commonly expressed antigens include osteocalcin, osteonectin, S-100 protein, SMA, NSE and CD99, but not CD34 [12].
We have observed only one case report of infantile fibrosarcoma (IFS) masquerading a sacrococcygeal teratoma [13]. Nearly all cases of IFS occur in the first year of life, 36-80% of those tumors are congenital with a slight male predominance. Histologically, this is a densely cellular neoplasm composed of intersecting fascicles of primitive round, oval, and spindle cells with a focal herringbone pattern. Mitotic activity may be prominent, hemangiopericytoma-like pattern may be seen. Immunophenotype features are nonspecific, but negative for CD34. IFS characterized by a chromosomal translocation, t (12; 15)(p13; q25),which involve ETV6 gene [2].
ConclusionTop
The extremely rare malignant SFT of the sacrum in 6-year-old child was described. A differential diagnosis is difficult and has to exclude both benign and malignant spindle cell bone lesions (hemangiopericytoma, spindle cell monophasic variant of SS, atypical histological variant of ES, PNST, solid variant of ABC, fibroblastic subtype of conventional osteosarcoma and IFS). A multidisciplinary approach (radiologist, surgeon, pathologist and etc.) is very helpful.
Conflict of interest
The authors declare no conflict of interest.
ReferencesTop
[1] Picci P, Manfrini M, Fabbri N, Gambarotti M, Vanel D. Atlas of Musculoskeletal Tumors and Tumorlike Lesions. The Rizzoli Case Archive, Springer International Publishing, Switzerland, 2014; pp.329–331.Article
[2] Fletcher CDM, Bridge JA, Pancras CW, Mertens HF. WHO Classification of tumors of soft tissue and bone. 4th Edition, IARC Press, 2013.
[3] Krishnan Unni K, Carrie Y Inwards. Dahlin’s Bone Tumors. Lippincott Williams & Wilkins, 2010 p.282.
[4] Petur Nielsen G, Andrew E Rosenberg, Vikram Deshpande, Francis J Hornicek, Susan V Kattapuram, Daniel I Rosenthal. Diagnostic Pathology Bone. 2013, pp.13,23.
[5] Son S, Lee SG, Jeong DH, Yoo CJ. Malignant solitary fibrous tumor of tandem lesions in the skull and spine. J Korean Neurosurg Soc. 2013; 54(3):246–249.Article Pubmed
[6] Liu Q, Liu J, Chen W, Mao S, Guo Y. Primary solitary fibrous tumors of liver: a case report and literature review. Diagn Pathol. 2013; 8:195.Article Pubmed
[7] Sharon W Wess, John R Goldblum. Soft Tissue Tumors. 5th Edition, 2008, pp.1120–1134.
[8] Verbeke SL, Fletcher CD, Alberghini M, Daugaard S, Flanagan AM, et al. A reappraisal of hemangiopericytoma of bone; analysis of cases reclassified as synovial sarcoma and solitary fibrous tumor of bone. Am J Surg Pathol. 2010; 34(6):777–783.Article Pubmed
[9] Mosquera JM, Flecher CD. Expanding the spectrum of malignant progression in solitary fibrous tumors: a study of 8 cases with a discrete anaplastic component – is this dedifferentiated SFT Am J Pathol. 2009; 33(9):1314–1321.Article Pubmed
[10] Fujikura T, Ishida M, Sekine K, Aoki H, Okubo K. Solitary Fibrous Tumor Arising from the Superior Nasal Turbinate: A Case Report. J Nippon Med Sch. 2012; 79(5):373–376.Article Pubmed
[11] Folpe AL, Goldblum JR, Rubin BP, Shehata BM, Liu W, et al. Morphologic and immunophenotypic diversity in Ewing family tumors: a study of 66 genetically confirmed cases. Am J Surg Pathol. 2005; 29(8):1025–1033.Article Pubmed
[12] Kubo T, Shimose S, Fujimori J, Arihiro K, Ochi M. Diversity of angiogenesis among mlignant bone tumors. Mol Clin Oncol. 2013; 1(1):131–136.Article Pubmed
[13] Al-Salem AH. Congenital-infantile fibrosarcoma masquerading as sacrococcygeal teratoma. J Pediatr Surg. 2011; 46(11):2177–2180.Article Pubmed
 Copyright
© 2012-2019 NobleResearch Group. All Rights Reserved
Copyright
© 2012-2019 NobleResearch Group. All Rights Reserved